174. Two remarks said that proposed Sec. 820.122 Storage really should be amended to be just like ISO 9001, and that the remainder of the necessities should be deleted and A part of a direction doc. A person comment said the term ``out of date'' need to be deleted mainly because, Whilst a device could not be marketed, thus which makes it obsolete, the factors for that machine should still be saved for buyer assist of the prevailing devices. FDA agrees that Sec. 820.122, now Sec. 820.150, may be a lot more per ISO 9001 and has revised the portion to harmonize with ISO 9001:1994. FDA hasn't deleted the time period ``out of date.'' FDA understands that a device may well no more be bought, but that elements and subassemblies should still be expected for consumer help; for that reason, Individuals elements or subassemblies usually are not ``obsolete.'' FDA's intent With this need is to ensure that only the suitable product or service be utilised or dispersed. FDA has deleted the prerequisite that Handle figures or identifications be legible and visual because it thinks the necessity is inherent in Sec.
Like other services in China, the Dalian plant was closed by the government for many days in early February, nevertheless it has operated given that then. Flamma decided to shut its Italian plant for one 7 days in March, “mostly to give men and women a split and commit time with their relatives,” Negrisoli states.
180. Several feedback underneath Sec. 820.a hundred and eighty Common requirements suggested that FDA delete the prerequisite that documents be stored to permit ``rapid retrieval'' simply because a reasonable time frame should be authorized. 1 comment said the wording from the portion needed to be amended to permit information to get Found in numerous areas, especially for overseas manufacturers and distributors. Two opinions stated the prerequisite must be competent by ``subject to conflicting legal prerequisites in other countries'' for the reason that some countries have ``blocking statutes'' that might prohibit the discharge of some facts. Just one comment said that anywhere the phrase ``all'' appeared in the necessities, FDA should get rid of it. FDA has rearranged this segment, and notes that data must be held in a very spot that's ``fairly available'' to both the producer and FDA investigators, and that information needs to be manufactured ``readily available.'' FDA expects that these documents will be produced accessible in the program of an inspection. In the event the foreign manufacturer maintains documents at remote destinations, these types of information can be predicted for being produced by the next Doing work working day or 2, at the most recent. FDA has clarified that data is often retained at besides the inspected institution, supplied that they're manufactured ``available'' for critique and copying. This could present foreign manufacturers and Original distributors the mandatory versatility. FDA hasn't experienced Sec. 820.180 in reaction for the comments to the ``blocking statues'' because if manufacturers choose to import medical equipment into America, then they need to comply with relevant statutory and regulatory demands, together with part 820. The documents section of this regulation is actually the same as that of the original CGMP and FDA has not discovered these ``blocking statutes'' to current a problem. Even more, international locations increasingly notice the value of a world marketplace, So FDA isn't going to foresee this problem to become a challenge in the future. In response on the comment on the phrase ``all'', FDA notes that in which a necessity exists for guaranteeing that records are preserved in a specific trend, a manufacturer ought to continue to keep all data topic towards the regulation in that way. The revised section helps make distinct that it's ``all records required'' from the regulation to which the area's demands pertain. 181. A few comments on Sec. 820.180(b), ``Report retention interval,'' mentioned that the segment ought to be amended mainly because all high-quality records will not be tied to a selected device; consequently, these excellent information might not need to be taken care of about the life span of a device. A few responses stated which the retention period of time necessity is unclear and burdensome, while some stated that the period of time really should be still left to the company to determine. Just one remark recommended the deletion of the necessities linked to photocopying documents in proposed Sec. 820.a hundred and eighty(b) as it is technology that isn't essentially getting used.
CGMP refers to the Present-day Excellent Manufacturing Follow restrictions enforced from the FDA. CGMP presents for units that assure right style and design, monitoring, and control of manufacturing procedures and facilities. Adherence for the CGMP laws assures the id, energy, quality, and purity of drug products by necessitating that manufacturers of medications adequately Manage manufacturing functions.
Less than the quality by style (QbD) paradigm, There exists a robust concentrate on approach comprehension of the affect of approach parameters and materials characteristics on products good quality. Applying process analytical engineering (PAT) gains such system know-how and develops riskbased quality control. In circulation method development, integrating in-line analytical systems presents a useful Device to comprehend and check the procedure in genuine time. Based on this analytical information and facts, method disorders can be optimized and taken care of throughout the Procedure; variations or difficulties might be determined and responded to instantly with out influencing downstream processes.six On top of that, developments in sensor technologies and procedure sampling can tremendously increase the capability of in-line checking and Manage.
“We've a number of disruptions in the provision chain, but not so intense. We've got most likely seen a little bit extra, especially this 7 days,” he informed C&EN in early April.
The raw content source chain is another critical component. Not only does the vendor need to be experienced, but they must also manage to make sure the very long-term well timed supply of needed raw substance quantities within the demanded quality.
7. Part 820.forty Doc Handle Some feedback thought that website the cost of implementing documentation techniques as well as other paperwork was understated. Nevertheless, ERG's estimates bundled the incremental compliance fees for formalizing a penned doc Management technique and ERG viewed as paperwork necessities in its estimation.
Availability of essential Uncooked materials has to be evaluated to make certain that These are readily available from current suppliers, or no matter whether new suppliers may be founded, to prevent a scenario where you are constrained by supply of the important Uncooked material or not able to import it.
linked deaths and critical injuries signify FDA's ideal projections, given the restrictions and uncertainties of the data and assumptions. The above mentioned numbers, having said that, never seize the quality of life losses to clients who working experience considerably less severe injuries than Those people described in MDR's, who practical experience nervousness because of cure having an unreliable healthcare system, or who practical experience inconvenience and extra clinical prices because of product failure.
a hundred and twenty. A couple of reviews said that proposed Sec. 820.sixty five Significant devices, traceability implies that traceability requirements exist for all devices. A number of other written feedback and oral testimony in the August and September 1995 conferences said that the wording of your Doing the job Draft was much too broad, obscure, and ambiguous, As well as in result would demand that all units be traced. As noted higher than, FDA has deleted the important system terminology. Section 820.sixty five is now entitled Traceability and uses the definition from the initial CGMP of a essential gadget to deliver the necessary clarity and delineation for this need. Hence, traceability is needed for the significant units stated inside the Federal Sign up recognize of March seventeen, 1988 (53 FR 8854). However, FDA is using the definition of significant device while in the need of Sec. 820.65, rather then a reference on the 1988 listing of critical units, for the reason that that list has not been updated considering that 1988 and there are no strategies to revise that checklist. Hence, it is very important that manufacturers make use of the definition throughout the prerequisite of Sec. 820.sixty five to ascertain if a specific device should be traced; it may not be enough to count solely within the 1988 listing. Manufacturers could uncover it advantageous to supply device, ton, or batch traceability for products for which traceability is not really a need to aid Regulate and limit the quantity of devices that may must be recalled due to defects or violations of your act. It is vital which the traceability specifications partially 820 will not be baffled Together with the Health care Unit Tracking regulation partially 821 (21 CFR aspect 821). The tracking regulation is meant in order that tracked equipment is often traced in the machine manufacturing facility to the person for whom the machine is indicated, that is definitely, the affected person. Helpful tracking of products in the manufacturing facility, from the distribution community (which include distributors, retailers, rental companies and also other industrial enterprises, machine user facilities, and certified practitioners) and, in the long run, to any person for whom the gadget is intended is essential for the efficiency of remedies prescribed with the act, including affected individual notification (segment 518(a) on the act (21 U.S.C. 360h(a)) or unit remember (portion 518(e).) In distinction, the traceability provision necessitates that a tool that meets the definition of the ``essential machine'' is often traced with the manufacturing facility only to the ``initial consignee'' as talked about in Sec.
two. Other Normal Responses Some manufacturers of low-threat gadgets plus some that have never experienced a product recall or MDR function questioned the merit and advantages of implementing style and design controls to all products. While in the proposed and ultimate CGMP regulation, FDA exempted Pretty much all class I products because the community health Gains acquired didn't exceed the costs of implementation. Nevertheless, FDA thinks that every one class II and III products really should be coated since their failure could adversely influence general public health. Even companies with superb previous documents put their customers at foreseeable future hazard if their style techniques are insufficient. ERG estimates that strict compliance to the final CGMP regulation will avert about 43 deaths and about 600 critical accidents every year.
With a chance to alter manufacturing volumes over the fly, organizations can improve resources and lessen squander, generating API manufacturing far more cost-powerful and environmentally sustainable.
It is also crucial that you take a look at the method from an environmental standpoint, making sure that all squander could be handled and disposed of appropriately, and also to make certain the procedure is scalable from laboratory through for the commercially projected scale.
Celebrity Then and Now
 Neve Campbell Then & Now!
Neve Campbell Then & Now!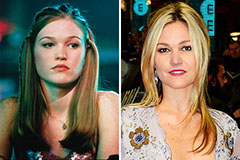 Julia Stiles Then & Now!
Julia Stiles Then & Now!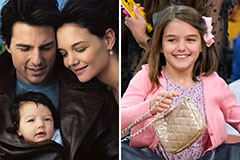 Suri Cruise Then & Now!
Suri Cruise Then & Now! Bo Derek Then & Now!
Bo Derek Then & Now! Ricky Schroder Then & Now!
Ricky Schroder Then & Now!